从原理上来讲,虚拟筛选可以分为两类,即基于受体(Receptor-based virtual screening)的虚拟筛选和基于配体(ligand-based virtual screening)的虚拟筛选。
基于受体的虚拟筛选也称为基于结构的虚拟筛选,利用分子对接技术,基于受体的三维结构,在结合位点处自动的匹配化合物数据库中的小分子,然后对可能的结合模式运用基于分子力场的打分函数进行结合能计算,最终得到化合物能量排名。
结构可以直接从PDB导入,也可以使用 File > Import Structures从工作目录中导入,本次教程以软件的实例进行基于结构的虚拟筛选(Structure-Based Virtual Screening)步骤演示。
①在工作目录中创建一个项目
在菜单栏点击File > Change Working Directory更改工作目录;在菜单栏点击File > Save Project As,更改文件名称为FXa_Glide,点击Save保存;
②导入预安装的文件
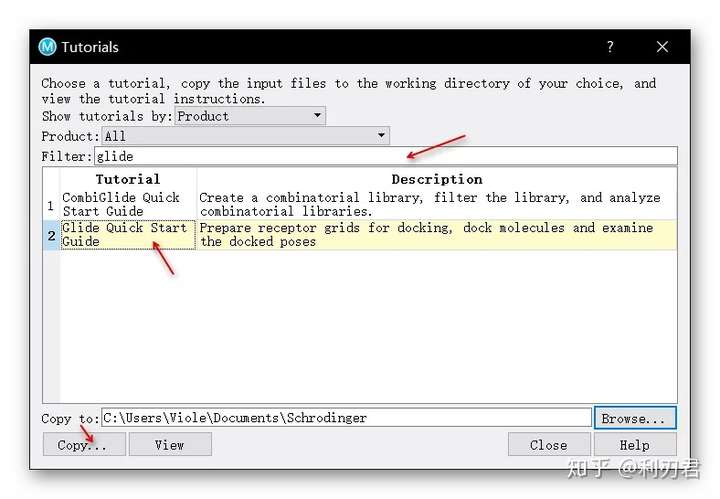
在菜单栏点击Help > Tutorials,在Filter一栏中输入Glide;
选择Glide Quick Start Guide,更改文件目录为设置好的工作目录;
点击Copy,此时所有的教程文件将复制到工作目录中;
点击Close关闭窗口;
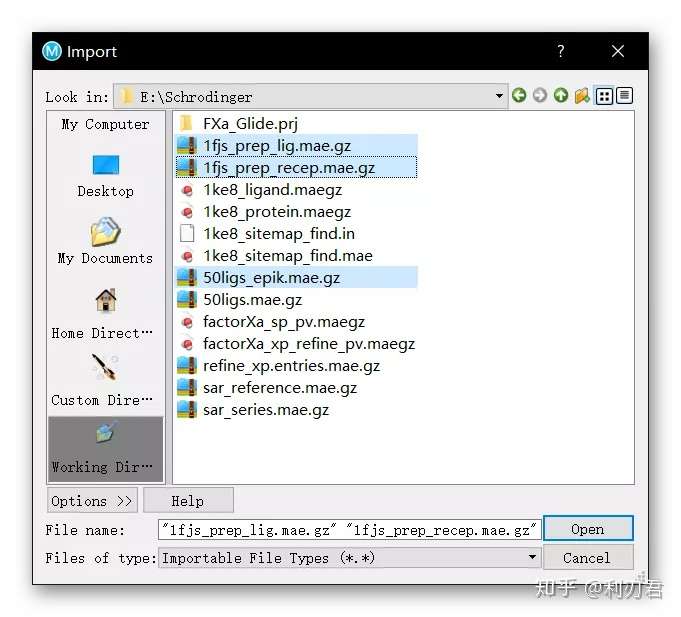
在菜单栏点击File > Import Structures;
选择文件1fjs_prep_lig.mae.gz, 1fjs_prep_recep.mae.gz,以及50ligs_epik.mae.gz,点击Open导入文件;
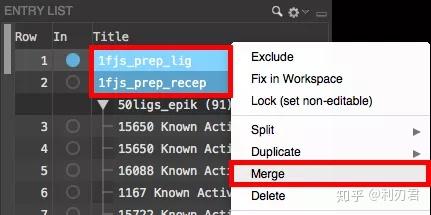
在项目列表中左键选择1fjs_prep_lig和1fjs_prep_recep;
鼠标右键选择Merge,此时两个项目将合并为一个;
双击产生的新项目,重命名为1fjs_prep_complex;
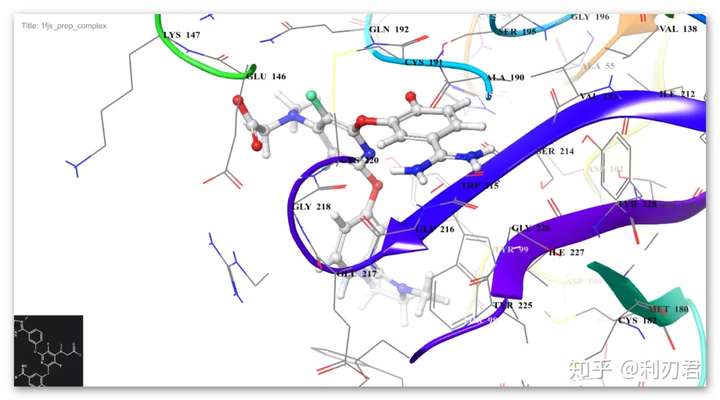
③定义结合位点
勾选1fjs_prep_complex旁边的圆圈以使其在显示窗口中显示;点击Presets,使1fjs_prep_complex 以预设方式显示;
在软件右上角点击Tasks > Browse > Receptor-Based Virtual Screening > Receptor Grid Generation,打开Receptor Grid Generation对话框;
在Receptor标签栏下的Define Receptor选项中, 检查Pick to Identify the ligand (Molecule)以及Show Markers是否勾选;
- 此时工作区弹出提示框提示单击配体中的一个原子。
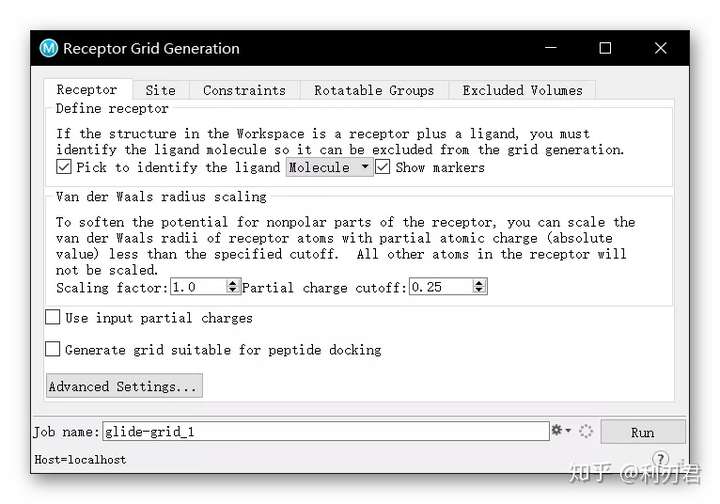
点击配体分子,此时在配体分子周围生成了一个紫色网格;
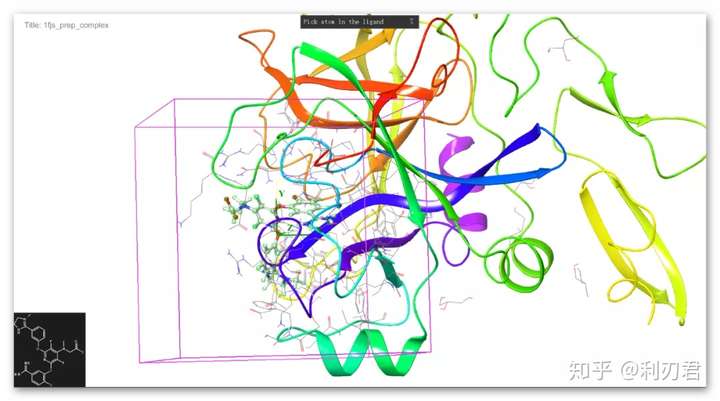
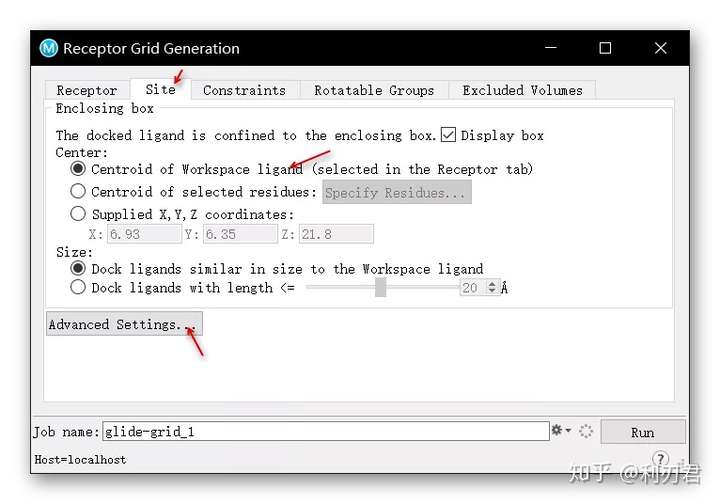
点击Site选项卡,选择Centroid of Workspace ligand (selected in the Receptor tab),即配体扩张法定义结合位点;点击Advanced Settings,此时出现一个绿色的网格;
- 绿色边界框定义了停靠分子的质心必须占据的区域。
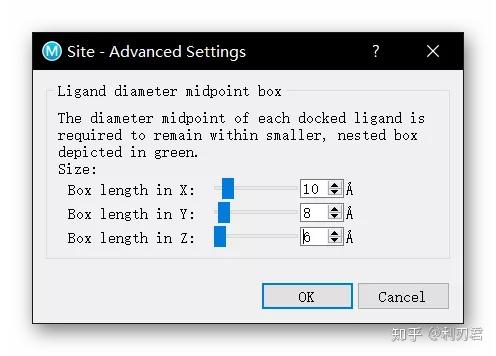
通过更改半径来改变绿色网格的大小,这里将X、Y和Z大小的设置分别调整为10、8和6埃;
单击OK确认;
④设置氢键约束
在快捷工具栏中,选择类型L使配体放大并居中显示;
在项目列表中,展开chain A,选择氨基酸残基ASP 189;点击Fit view to selected atoms;
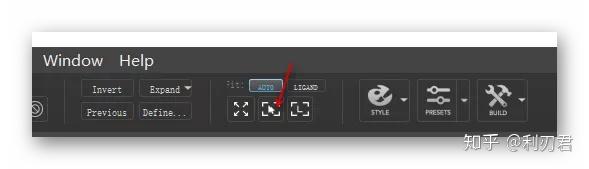
在Receptor Grid Generation对话框中, 选择Constraints选项卡;选择H-bond/Metal (0)选项卡;
- 此时工作窗口中提示选择受体原子作为约束;
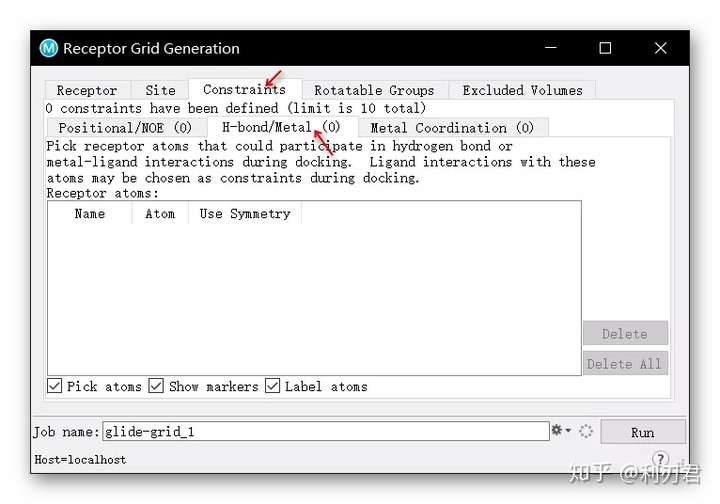
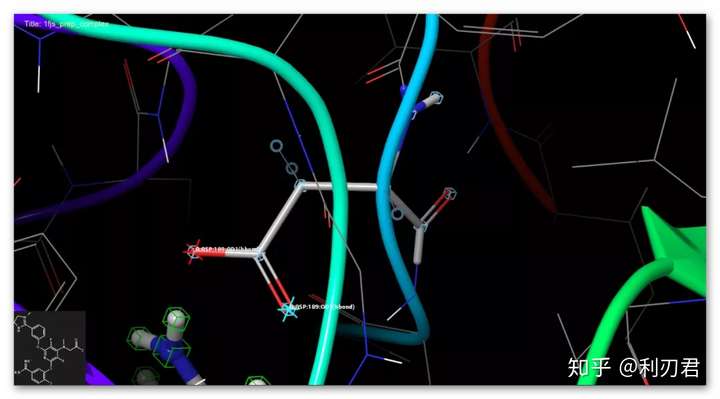
点击ASP189残基上的氧原子,此时残基上的两个氧都突出显示;氢键约束在受体原子表中被定义;
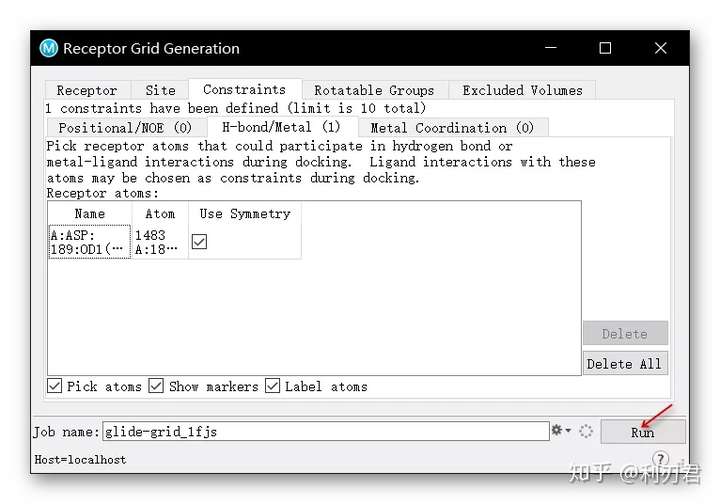
将任务名称更改为glide-grid_1fjs,单击“Run”运行任务;
这任务大约需要一分钟,一个名为glide-grid_1fjs的文件夹被写入工作目录。
以上就是利用借助Schrodinger软件进行基于结构的虚拟筛选的教程上篇,下篇教程将为大家分享分子对接及结果分析的教程~